Usage¶
There are two search modes, specified by the –mode argument. By default (i.e. –mode not specified), it will be set to remote, and a fully remote cblaster search will begin using NCBI’s BLAST API. Alternatively, local mode performs a search against a local diamond database, which is much quicker (albeit requiring some initial setup).
Performing a remote search via the NCBI BLAST API¶
At a minimum, a search could look like one of the following:
$ cblaster search -qf query.fasta
$ cblaster search -qi QBE85649.1 QBE85648.1 QBE85647.1 QBE85646.1 ...
This will launch a remote search against the non-redundant (nr) protein database, retrieve and parse the results, then report any blocks of hits to the terminal. By default, hits are only reported if they are above 30% percent identity and 50% query coverage, and have an e-value below 0.01. If we wanted to be stricter, we could change those values with the following:
$ cblaster search -qf query.fasta --min_identity 70 --min_coverage 90 --evalue 0.001
You can also pass in NCBI search queries using -eq / –entrez_query to pre-filter the target database, which can result in vastly reduced run-times and more targeted results. For example, to only search against Aspergillus sequences:
$ cblaster search -qf query.fasta --entrez_query "Aspergillus"[ORGN]
Look here for a full description of Entrez search terms.
Searching a local database using DIAMOND¶
Alternatively, a local DIAMOND database can be searched by specifying:
$ cblaster search -qf query.fasta --mode local --database db.dmnd
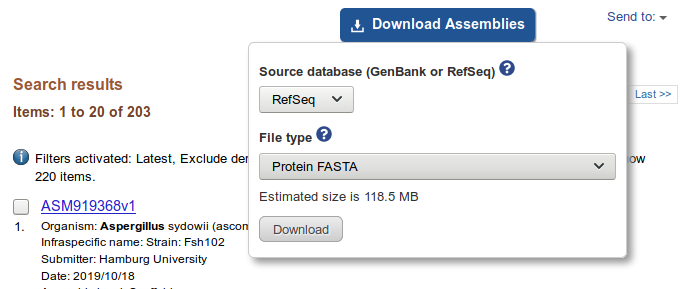
For this to work, the database must consist of sequences derived from NCBI, such that their identifiers can be used for retrieval of sequences/genomic context. The easiest way to set this up is via NCBI’s batch assembly download option. For example, to build a database of Aspergillus protein sequences:
- Search the NCBI Assembly database for Aspergillus genomes
- Click ‘Download Assemblies’, select ‘Protein FASTA’ and click ‘Download’
- Extract all FASTA files and concatenate them
$ pigz -d *.gz
$ cat *.faa >> proteins.faa
- Build the DIAMOND database
$ diamond makedb --in proteins.faa --db proteins
...
$ ls
database.faa
database.dmnd
```
- Run cblaster against the newly created databse
$ cblaster search -m local -qf query.fa -db database.dmnd <options>
Alternatively, you could use ncbi-genome-download to retrieve the sequences from the command line.
Performing a fully local search¶
cblaster can also perform fully local searches, forgoing the need for any interaction with NCBI whatsoever. To do this, cblaster builds a faux-database JSON file from a list of GenBank files, and generates a diamond database of all protein sequences in this database. Then, cblaster searches can be run against the created diamond database, and genomic context obtained from the JSON file. For example:
- Build diamond and JSON databases
$ cblaster makedb path/to/folder/*.gbk mydatabase
[12:06:32] INFO - Parsing 3 files...
[12:06:32] INFO - 1. /path/to/folder/one.gbk
[12:06:33] INFO - 2. /path/to/folder/two.gbk
[12:06:34] INFO - 3. /path/to/folder/three.gbk
[12:07:21] INFO - Writing FASTA file with database sequences: mydatabase.faa
[12:07:21] INFO - Building DIAMOND database: mydatabase.dmnd
[12:07:36] INFO - Building JSON database: mydatabase.json
Now, everything should be in the folder:
$ ls
mydatabase.faa
mydatabase.dmnd
mydatabase.json
Note that although this produces a FASTA file with all protein sequences in the database, the file is NOT actually required for searches. It is merely created for the purpose of building the diamond database, and can easily be recreated by loading the JSON database in code as a database.DB instance and running write_fasta() on an open file handle.
- Run a cblaster search against the built databases
$ cblaster search -qf query.fasta --json mydatabase.json -db mydatabase.db <options>